Molecular Modeling Practical
This tutorial introduces the student to the practice of Molecular Dynamics (MD) simulations of proteins. The protocol used is a suitable starting point for investigation of proteins, provided that the system does not contain non-standard groups. At the end of the tutorial, the student should know the steps involved in setting up and running a simulation, including some reflection on the choices made at different stages. Besides, the student should know how to perform quality assurance checks on the simulation results and have a feel for methods of analysis to retrieve information.
Molecular Dynamics Hands-on Session
Molecular dynamics (MD) simulations consist of three stages: First, the input data (the simulation system) have to be prepared. Second, the production simulation can be run and finally the results have to be analysed and be put in context. Although the second stage is the most computationally demanding step, with simulations commonly running for several months, the most laborious steps are the preparation and the analysis. This part of the tutorial provides an example session of setting up a protein for molecular dynamics simulation, running it and analysing the results.
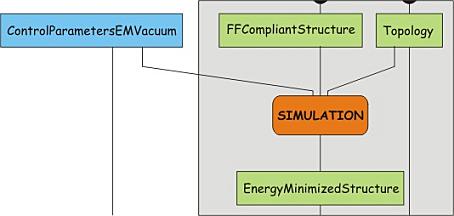
This tutorial uses Gromacs (http://www.gromacs.org) version 4.5.3 and most of the commands given are specific to this package. The workflow(s) will be more general though and will also apply to other MD packages. Each step will be presented schematically, as a node with input and output. A step can be the execution of a single program or a combination of programs, with output from one being directed to the other. In the case of a program, the input/output types and flags will be specified. For most of the programs/steps only the input/output options used are listed. Most of the programs offer options for more advanced control, which will not be mentioned here. For those interested, the help page for each program gives a full listing and description of the functionality. The help page can be shown by running a program with the option "-h". In the workflow, programs are indicated by the name on a yellow background. Input/output data is given as a descriptive name on a green background. The blue blocks indicate input data which is essentially not dependent on the structure under study. The orange blocks are processes comprising of several steps in terms of the programs involved. In the present scheme, orange blocks are solely used for the simulation steps, which are explained in more detail below. The lines connect the data to the processes and for each program the command line option for the input or the output is indicated in white on black.
{kind=link}
Preparing the system
The preparation of the simulation system is the most important stage of MD. MD simulations are performed to gain insight into processes on an atomistic scale. They can be used to understand experimental observations, to test hypotheses or as a basis for new hypotheses to be assessed experimentally. However, for each of these cases, the simulations have to be designed such that they are suited for the purpose. This means that the setup should be performed with care.
Missing residues, missing atoms and non-standard groups
In this tutorial we consider the simulation of a protein. The first step in the sequence then is the selection of a starting structure, which has been covered on the previous page. The second step should be checking whether the structure has missing residues and atoms, which should be modeled in some way. In our case, the structures are complete and this step is skipped.
Another problem which one should be aware of is that structure files may contain non-standard residues, modified residues or ligands, for which force field parameters are not yet available. Such groups should either be removed or parameters should be determined for them, which is an advanced topic in MD. The structures in this tutorial only consist of natural amino acids.
Structure quality
It is good practice to run further checks on the structure to assess the quality. For instance, the refinement process in crystal structure determination may not always yield proper orientations of glutamine and asparagine amide groups. Also, the protonation state and side chain orientation of histidine residues may be problematic. A listing of the most relevant chemical properties of the proteinogenic aminoacids is available on Wikipedia. To perform a proper validation of the structure, several programs and servers exist (e.g. WHATIF). For this tutorial we will assume that the structure is good enough for our purpose and continue with preparing the simulation system.
Structure conversion and topology
 A molecule is defined by the coordinates of the atoms as well as by a
description of the bonded and nonbonded interactions. Since the structure
obtained from the PDB only contains coordinates, we first have to construct the
topology, which describes the system in terms of atom types, charges, bonds,
etc. This topology is specific to a certain force field and the force field to
be used is one of the issues requiring careful consideration. Here we use the
GROMOS96 53a6 united atom force field, which is parameterized to give good
predictions of free energies of solvation of amino acid side chains and which
generally gives good agreement with NMR experiments.
A molecule is defined by the coordinates of the atoms as well as by a
description of the bonded and nonbonded interactions. Since the structure
obtained from the PDB only contains coordinates, we first have to construct the
topology, which describes the system in terms of atom types, charges, bonds,
etc. This topology is specific to a certain force field and the force field to
be used is one of the issues requiring careful consideration. Here we use the
GROMOS96 53a6 united atom force field, which is parameterized to give good
predictions of free energies of solvation of amino acid side chains and which
generally gives good agreement with NMR experiments.
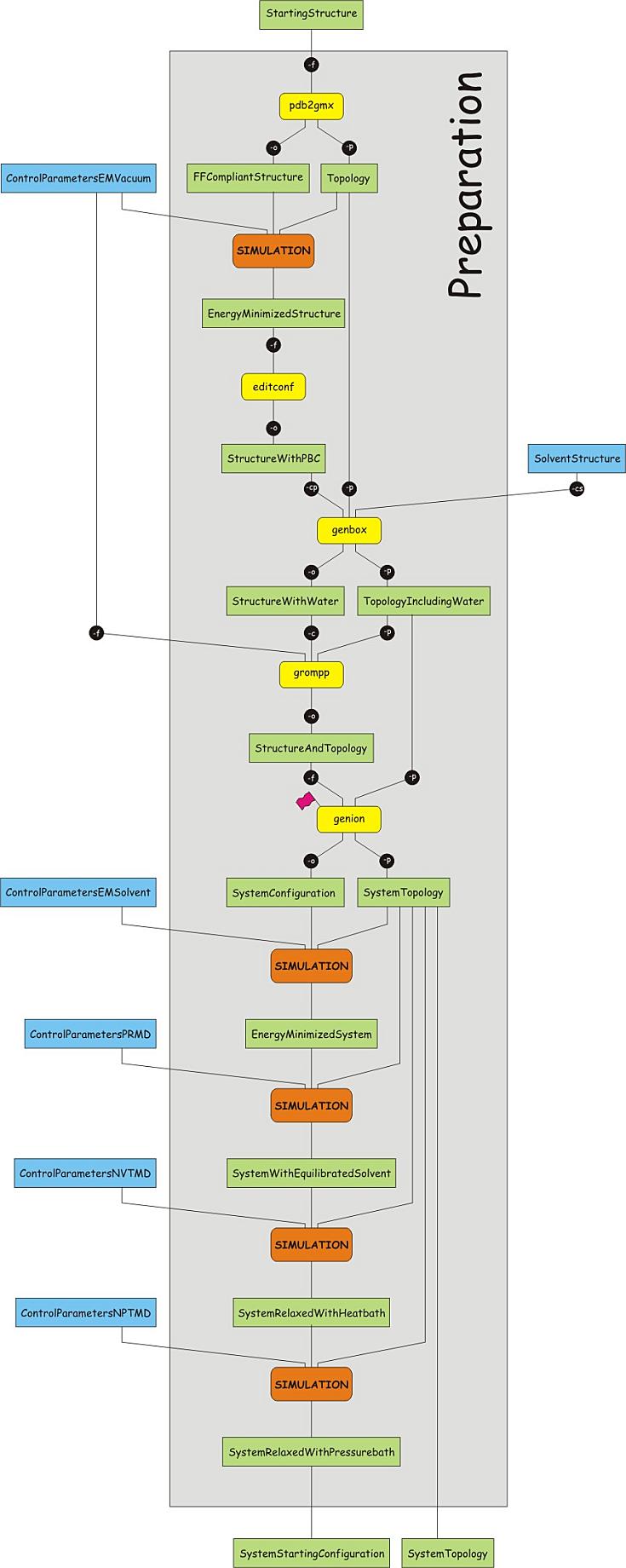
It is important that the topology matches with the structure, which means that
the structure needs to be converted too, to adhere to the force field used. To
convert the structure and construct the topology, the program pdb2gmx can be
used. This program is designed to build topologies for molecules consisting of
distinct building blocks, such as amino acids. It uses a library of building
blocks for the conversion and will fail to recognize molecules or residues not
present in the library. Issue the following command to convert the structure;
choose the GROMOS 53a6 force field and the SPC water model when prompted. Note the flag -ignh, which
causes hydrogen atoms present in the file to be removed, and to be rebuilt
according to the description in the force field.
pdb2gmx -f protein.pdb -o protein.gro -p protein.top -ignh
You can always generate a pdb file from a gro file using the
following command:
editconf -f XXXXX.gro -o XXXXX.pdb
Read through the output on the screen and check the choices made for the histidine protonation and the resulting total charge of the protein. Also browse through the input structure file (protein.pdb, pdb format) and output structure file (protein.gro, gromacs format). Note the differences between the two formats. Browse then through the topology file and look at its structure.
Write down the number of atoms before and after the conversion and try to explain the difference
List the atoms, atom types and charges from a tyrosine residue as given in the topology file
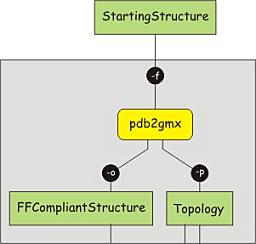
Energy minimization of the structure (vacuum)
 Now
a structure is generated in the correct format for the force field chosen,
as well as the corresponding topology. However, the conversion of the
structure involves the deletion and or addition of hydrogen atoms and may
cause strain to be introduced, e.g. due to atoms positioned too close
together. Therefore, we have to perform an energy minimization on the
structure, and let it relax a bit. This is in fact a simulation step, and involves two
processes. First, the structure and the topology are combined into a
single description of the system, together with a number of control
parameters. This yields a run input file, which can be used as the single
input for the simulation program mdrun. The advantage of splitting the two
sub-processes is that the run-input file can be transferred to a dedicated
(high-performance) computer network or a supercomputer and be processed
there. However, this is only relevant for the production simulation.
Now
a structure is generated in the correct format for the force field chosen,
as well as the corresponding topology. However, the conversion of the
structure involves the deletion and or addition of hydrogen atoms and may
cause strain to be introduced, e.g. due to atoms positioned too close
together. Therefore, we have to perform an energy minimization on the
structure, and let it relax a bit. This is in fact a simulation step, and involves two
processes. First, the structure and the topology are combined into a
single description of the system, together with a number of control
parameters. This yields a run input file, which can be used as the single
input for the simulation program mdrun. The advantage of splitting the two
sub-processes is that the run-input file can be transferred to a dedicated
(high-performance) computer network or a supercomputer and be processed
there. However, this is only relevant for the production simulation.
{kind=link}
To perform these steps first save the parameter file minim.mdp to your directory. This file contains the control parameters for the energy minimization. Have a look at the contents of this file. Notice the line starting with "integrator", which specifies the algorithm to be used. In this case it is set to perform a steepest descent energy minimization of 5000 steps. Now, combine the structure, topology and control parameters using grompp:
grompp -v -f minim.mdp -c protein.gro -p protein.top -o protein-EM-vacuum.tpr
grompp will note that the system has a non-zero charge and will print some other information regarding the system and the control parameters. The program will also generate an additional output file, which contains the settings for all control parameters (mdout.mdp). Now, continue with mdrun:
mdrun -v -deffnm protein-EM-vacuum -c protein-EM-vacuum.gro
Since there are only about 1500 atoms, the energy minimization finishes quickly. The -v flag causes the potential energy and the maximum force to be printed at each step, allowing following the minimization. The -deffnm flag sets the default filename for all file options, it avoids multiple flags usage. Now that the structure is relaxed, it is time to set up the periodic boundary conditions and add the solvent.
Which method was used for the energy minimization?
How many steps were specified in the parameter file and how many steps did the minimization take?
What could cause the energy minimization to stop before the specified number of steps was reached?
What is the final potential energy of the system?
Compare the structures before and after energy minimization by loading them into PyMol
Periodic boundary conditions
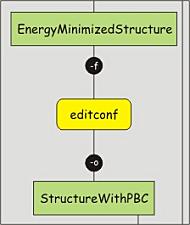 Before adding the solvent, a general layout (the space) of the simulation setup
has to be chosen. Most commonly simulations are performed under periodic
boundary conditions (PBC), meaning that a single unit cell is defined, which can be
stacked in a space filling way. In this way, an infinite, periodic system can
be simulated, avoiding edge effects due to the walls of the simulation
volume. There are only a few general shapes available to set up PBC. We will
use a rhombic dodecahedron
because it corresponds to the optimal packing of a
sphere, and is therefore the best choice for freely rotating
molecules. To disallow direct interactions between periodic images we set a
minimal distance of 1.0 between the protein and the wall of the cell, such that
two neighbours will not be closer than 2.0 nm. Periodic boundary conditions are
set with editconf:
Before adding the solvent, a general layout (the space) of the simulation setup
has to be chosen. Most commonly simulations are performed under periodic
boundary conditions (PBC), meaning that a single unit cell is defined, which can be
stacked in a space filling way. In this way, an infinite, periodic system can
be simulated, avoiding edge effects due to the walls of the simulation
volume. There are only a few general shapes available to set up PBC. We will
use a rhombic dodecahedron
because it corresponds to the optimal packing of a
sphere, and is therefore the best choice for freely rotating
molecules. To disallow direct interactions between periodic images we set a
minimal distance of 1.0 between the protein and the wall of the cell, such that
two neighbours will not be closer than 2.0 nm. Periodic boundary conditions are
set with editconf:
editconf -f protein-EM-vacuum.gro -o protein-PBC.gro -bt dodecahedron -d 1.2
Have a look at the output of editconf, and note the changes in the
volume. Also, have a look at the last line of the file protein-PBC.gro
 . In the gromacs format (.gro), the last line specifies
the unit cell shape. It always uses the triclinic matrix representation, with
the first three numbers specifying the diagonal elements (xx, yy, zz) and the last six
giving the off-diagonal elements (xy, xz, yx, yz, zx, zy).
. In the gromacs format (.gro), the last line specifies
the unit cell shape. It always uses the triclinic matrix representation, with
the first three numbers specifying the diagonal elements (xx, yy, zz) and the last six
giving the off-diagonal elements (xy, xz, yx, yz, zx, zy).
What is the new unit cell volume?
Solvent addition
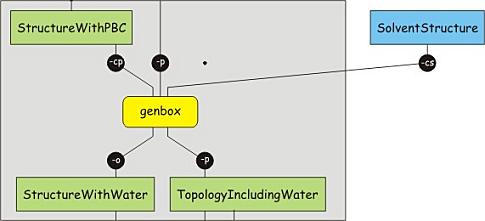 Now that the unit cell is set up, solvent can be added. There are several
solvent models, each of which is more or less intimately linked to a force
field. The GROMOS96 force fields are generally used with the simple point
charge (SPC) water model, which you should have chosen before. The topology is not required for solvent generation,
but may be updated to include the solvent added. To fill the unit cell with SPC issue the following
command:
Now that the unit cell is set up, solvent can be added. There are several
solvent models, each of which is more or less intimately linked to a force
field. The GROMOS96 force fields are generally used with the simple point
charge (SPC) water model, which you should have chosen before. The topology is not required for solvent generation,
but may be updated to include the solvent added. To fill the unit cell with SPC issue the following
command:
genbox -cp protein-PBC.gro -cs spc216.gro -p protein.top -o protein-water.gro
Have a look at the end of the file protein.top to see the addition, and check the numbers with the amount of solvent added according to genbox.
How many water molecules are added to the system and what volume does that correspond to?
How many atoms are in the system now?
Transform the .gro file in a .pdb file with the command given before. Load the solvated structure (protein-water.pdb) in Pymol. In Pymol, the unit cell can be visualized using the command
show cell
This draws a wireframe, showing the edges of the triclinic unit cell. Although it may not be obvious, this triclinic shape corresponds to a rhombic dodecahedron. Another thing to note is that the solvent is not all inside the unit cell, but is arranged as a rectangular brick. Quite likely, the protein is partially sticking out of the water.
Why is it not a problem to have the protein sticking out of the water?
Addition of ions: counter charge and concentration
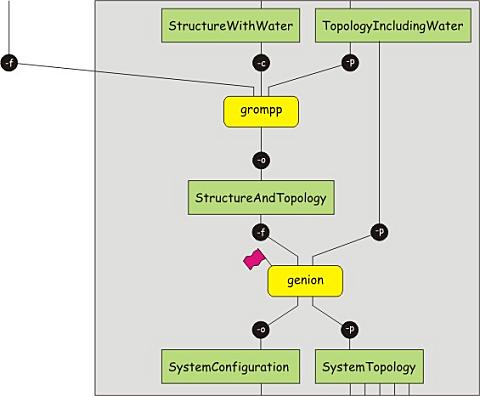 At this point, we have a solvated protein, but the net charge of the system
still remains. To make the system neutral we have to add a number of
counterions. In addition, it may be considered good practice to add ions up to
a certain concentration. The program genion can take care of both tasks, but
requires a run input file as input, i.e. a file containing both the structure
and the topology. As we saw before, such a file can be generated with grompp:
At this point, we have a solvated protein, but the net charge of the system
still remains. To make the system neutral we have to add a number of
counterions. In addition, it may be considered good practice to add ions up to
a certain concentration. The program genion can take care of both tasks, but
requires a run input file as input, i.e. a file containing both the structure
and the topology. As we saw before, such a file can be generated with grompp:
grompp -v -f minim.mdp -c protein-water.gro -p protein.top -o protein-water.tpr
Subsequently, the file protein-water.tpr can be used as input for genion. We specify a concentration of 0.15 M (-conc 0.15) of NA+/CL- (-pname NA+ -nname CL-) to be added and indicate that an excess of one species of ion has to be added to neutralize the system (-neutral). genion will ask for a group of molecules to be used to partly replace with ions. The group 'SOL' should be chosen.
genion -s protein-water.tpr -o protein-solvated.gro -conc 0.15 -neutral -pname NA+ -nname CL-
Note the numbers of ions added, and verify that an excess of chloride ions is added for neutralization. Having replaced a number of water molecules with ions, the system topology in protein.top is not correct anymore.
Edit the topology file and decrease the number of solvent molecules. Also add a line specifying the number of NA ions and a line specifying the amount of CL. NOTE that different versions of gromacs may use different names for the ions, either NA+ and CL- or NA and CL. If one gives trouble, try the other!
How many sodium and chloride ions are added to the system?
Energy minimization of the solvated system
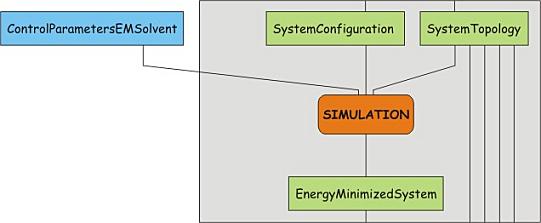 Now the whole simulation system is defined. The only steps before starting a
production run involve the controlled relaxation of the system. The generation
of solvent as well as the placement of ions, may have caused unfavourable
interactions, e.g. overlapping atoms, equal charges too close
together. Therefore, the system is energy minimized again, following the same
steps as before run grompp and mdrun:
Now the whole simulation system is defined. The only steps before starting a
production run involve the controlled relaxation of the system. The generation
of solvent as well as the placement of ions, may have caused unfavourable
interactions, e.g. overlapping atoms, equal charges too close
together. Therefore, the system is energy minimized again, following the same
steps as before run grompp and mdrun:
grompp -v -f minim.mdp -c protein-solvated.gro -p protein.top -o protein-EM-solvated.tpr
mdrun -v -deffnm protein-EM-solvated -c protein-EM-solvated.gro
How many steps were specified in the parameter file and how many steps did the minimization take?
What is the final potential energy of the system?
Relaxation of solvent and hydrogen atom positions: Position restrained MD
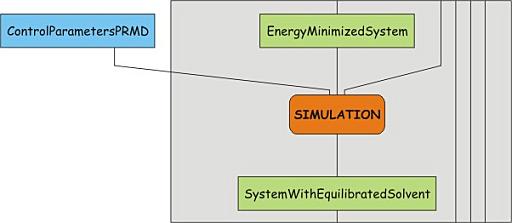 With the greatest strain dissipated from the system, we now let the solvent
adapt to the protein, i.e. we allow the solvent to move freely, while keeping
the non-hydrogen atoms of the proteins more or less fixed to the reference
positions. This is done to assure that the solvent configuration 'matches' the
protein. This step is the first actual MD step. The control parameters for this
step can be found in pr.mdp, which should be
downloaded. Have a look at this file and note the integrator used as well as
the define statement. The latter is used to allow flow control in the topology
file. define = -DPOSRES will cause the keyword "POSRES" to be globally
defined. Have a look at the end of the topology file to see how this is used to
turn on position restraints. Use grompp and mdrun to run the simulation:
With the greatest strain dissipated from the system, we now let the solvent
adapt to the protein, i.e. we allow the solvent to move freely, while keeping
the non-hydrogen atoms of the proteins more or less fixed to the reference
positions. This is done to assure that the solvent configuration 'matches' the
protein. This step is the first actual MD step. The control parameters for this
step can be found in pr.mdp, which should be
downloaded. Have a look at this file and note the integrator used as well as
the define statement. The latter is used to allow flow control in the topology
file. define = -DPOSRES will cause the keyword "POSRES" to be globally
defined. Have a look at the end of the topology file to see how this is used to
turn on position restraints. Use grompp and mdrun to run the simulation:
Remark: edit the file pr.mdp and change the value of gen_seed
grompp -v -f pr.mdp -c protein-EM-solvated.gro -p protein.top -o protein-PR.tpr
mdrun -v -deffnm protein-PR
What is the length of the simulation in picoseconds?
How is the inclusion of the position restraints handled in the topology file?
It is interesting to have a look at what happens with the total, potential
and kinetic energies. In the previous steps, the particles did not have
velocity, so there was no kinetic energy and hence no temperature. Now, at
the start of the simulation, the atoms were assigned velocities and with
that got kinetic energy.
The energy information from the simulation is stored in an unreadable
(binary) file format with extension .edr. The information in this file can
be extracted using the gromacs tool g_energy. Plot the temperature and the
potential, kinetic and total energy of the system.
NOTE: You can run the following commands and answer the questions while the next simulation step is running and save some time. If this doesn't make sense, just continue with the commands here. Otherwise, run grompp and mdrun for the simulation step below and run the following commands in a separate terminal window, but in the right directory.
g_energy -f protein-PR.edr -o thermodynamics-PR.xvg
This will present you a list of energy terms you can select for writing to
the file given for output. Find the numbers that corresponds to the
Temperature, Potential Energy, Kinetic Energy, and Total Energy and enter them. Finally type a 0 (zero) and press enter again
The output files (.xvg) can be viewed with the program 'xmgr' or 'xmgrace', one of which should be installed. Alternatively, these files can be viewed with the Python script xvg2ascii.py, which draws the graphs in the terminal window.
xmgrace -nxy thermodynamics-PR.xvg
Note that the green curve is the total energy, the black one is the potential, the blue one is the temperature and finally the red one is the kinetic energy. You can change the legend by clicking on the curve and filling in the 'Legend' box. Don't forget to 'accept' if you want to apply the changes.
What happens with the temperature?
What happens to the potential/kinetic/total energy and how can this be explained?
Unrestrained MD simulation, turning on temperature coupling
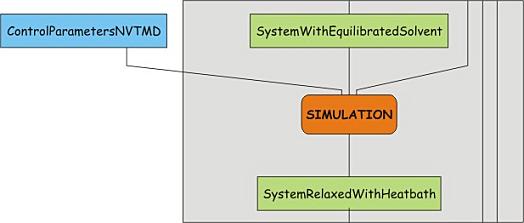 The system should now be relatively free of strain. Time to turn up the heat,
and start to couple the system to the heat bath. In other words, a short run
will be performed in which the temperature coupling is turned on and the system
is allowed to relax to the new conditions. Download the control parameter file
nvt.mdp and have a look at the temperature coupling
parameters in it. Then run the simulation, using grompp and mdrun:
The system should now be relatively free of strain. Time to turn up the heat,
and start to couple the system to the heat bath. In other words, a short run
will be performed in which the temperature coupling is turned on and the system
is allowed to relax to the new conditions. Download the control parameter file
nvt.mdp and have a look at the temperature coupling
parameters in it. Then run the simulation, using grompp and mdrun:
grompp -v -f nvt.mdp -c protein-PR.gro -p protein.top -o protein-NVT.tpr
mdrun -v -deffnm protein-NVT
There are two groups which are separately coupled to a heat bath. Which groups are that and what do you think is in each of these groups?
After the run finishes have a look at the energies and temperature. Extract them in the same way as before. Note again that you may want to start the next simulation before looking at the energies from this one.
g_energy -f protein-NVT.edr -o thermodynamics-NVT.xvg
Have a look at the energy graphs.
What happens to the temperature?
What happens to the potential/kinetic/total energy and how can this be explained?
Unrestrained MD simulation, turning on pressure coupling
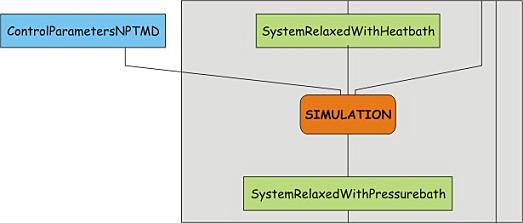 After relaxation of the temperature and the temperature coupling, it is time to
start putting things under pressure. Perform a short simulation with pressure
coupling turned on, using the control parameter file
npt.mdp. Have a look at this file and identify the
parameters controlling the pressure coupling.
After relaxation of the temperature and the temperature coupling, it is time to
start putting things under pressure. Perform a short simulation with pressure
coupling turned on, using the control parameter file
npt.mdp. Have a look at this file and identify the
parameters controlling the pressure coupling.
Also note that we reassign velocities to the particles, which is
controlled with the parameter gen_vel. Below that line, the temperature
for the distribution of velocities (the Maxwell distribution) is set and
the seed for the random number generator is specified (gen_seed). gen_seed
is currently set to 9999, but should be changed to a new value.
MD simulations are deterministic, so starting with the same set of
coordinates and velocities and the same parameters would cause the
outcomes of the simulations to be identical, which is not what we
want.
After you've edited the parameter file prepare and run the simulation:
Remark: edit the file npt.mdp and change the value of gen_seed
grompp -v -f npt.mdp -c protein-NVT.gro -p protein.top -o protein-NPT.tpr
mdrun -v -deffnm protein-NPT
After the run finishes, again have a look at the energies and the temperature. Also have a look at the pressure. Extract all of them in the same way as before.
g_energy -f protein-NPT.edr -o thermodynamics-NPT.xvg
Have a look at the energy graphs, the temperature and the pressure.
What happens to the temperature?
What happens to the pressure?
The last question relates directly to the extraction of thermodynamic properties from systems consisting of limited numbers of particles. Thermodynamics is, crudely spoken, about the behaviour of large collections of particles; billions rather than thousands. Averaging a properties over many many particles will yield values which have small fluctuations. On the other hand, averaging over a small number of particles inevitably gives large fluctuation.
Since we now do pressure coupling, the density of the system may also change. Extract the density from the energy file too:
g_energy -f protein-NPT.edr -o density-NPT.xvg
How does the density behave over time?
Why does the density of the system change if pressure coupling is turned on?
Production Simulation
Now finally, we have a more or less equilibrated solvated system containing our protein of interest and it is time to start a production simulation. Mind that production simulation does not imply that the whole simulation can be used for analysis of the properties of interest. Although some of the memory of the initial configuration has been erased, it is unlikely that the system has already reached equilibrium. In the analysis section we will investigate which part of the simulation can be considered in equilibrium and is thus suited for further processing. But first it is time to set up the simulation. This merely involves running another simulation step, similar to the last step of the preparation. However, this is another of these times where it is necessary to think about the purpose of the simulation. The control parameters have to be chosen such that the simulation will allow analysing the properties one has in mind. Questions to be addressed are:
- At what time scale do the processes of interest manifest themselves or how long should the simulation be run?
- How many frames are necessary, or what time resolution is required?
- Is there need to store velocities?
- Should all atoms be in the output, or is it sufficient to save only protein coordinates?
- What frequency should be used for writing energies and log entries?
- What frequency should be used for writing check point coordinates and velocities?
- ...
We're going to try and run 1 nanosecond simulations.
Download or copy the control parameter file md.mdp and
have a look at its contents.
How many steps are needed to get a total simulation time of 1 ns?
You'll have to edit the file and
specify how many steps should be taken to get a total simulation time of
1 ns. Then take the final structure file and topology resulting from
the preparation and combine them into a run input file using grompp.
grompp -v -f md.mdp -c protein-NPT.gro -p protein.top -o topol.tpr
And run the simulation
mdrun -v
While the simulation is running you can take the time to review the several steps that brought us to this point. Go through each mdp file noting the differences. It can also be didactic to understand what sort of information is stored in run input (.tpr) files. These are in a binary format, and it is not possible to directly have a look at the contents. The program to make the contents of the run input file readable is gmxdump. The output usually spans many pages and therefore it is advisable to redirect the output to file or to 'more' or 'less', which allow browsing the content. Issue the following command and inspect the structure of the output (replace the name topol.tpr with whaterver .tpr file you're inspecting):
gmxdump -s topol.tpr | less