


























































































































































Dimerization of GPCRs
- Details
- Created: Thursday, 07 June 2012 23:00
- Last Updated: Saturday, 09 November 2013 13:13
First report on the free energy surface of membrane protein dimerization
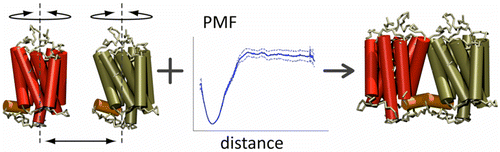
Abstract: G protein-coupled receptors (GPCRs) self-assemble into supramolecular structures in native bilayers, but the structural determinants of receptor oligomerization are not known. We carried out multiple self-assembly coarse-grained molecular dynamics (CGMD) simulations of model membranes containing up to 64 molecules of the visual receptor rhodopsin over time scales reaching 100 μs. The simulations show strong preferential interaction modes between receptors. Two primary modes of receptor–receptor interactions are consistent with umbrella sampling (US)/potential-of-mean-force (PMF) calculations as a function of the distance between a pair of receptors. The preferential interfaces, involving helices (H) 1/8, 4/5 and 5, present no energy barrier to forming a very stable receptor dimer. Most notably the PMFs show that the preferred rhodopsin dimer exists in a tail-to-tail conformation, with the interface comprising transmembrane H1/H2 and amphipathic H8 at the extracellular and cytoplasmic surfaces, respectively. This dimer orientation is in line with earlier EM, X-ray, and crosslinking experiments of rhodopsin and other GPCRs. Less stable interfaces, involving H4 and H6, have a free energy barrier for desolvation (delipidation) of the interfaces and appear to be designed to stabilize “lubricated” (i.e., lipid encased) dimers. The overall CGMD strategy used here is general and can be applied to study the homo- and hetero-dimerization of GPCRs and other transmembrane proteins. Systematic extension of the work will deepen our understanding of the forces involved in the membrane organization of integral membrane proteins.