martinize
- Details
-
Last Updated: Thursday, 17 August 2017 11:59
Martinize is a python script to generate Martini protein topology and structure files based on an atomistic structure file. It replaces the old seq2itp, atom2cg and ElNeDyn scripts. The produced topology and structure files are in a format suitable for Gromacs.
The current version (2.x) has been used rather extensively, however it might still contain errors or bugs. Any feedback is more than welcome! The script is "concatenated": all the different classes, modules and forcefields are in one file. If you want to make changes yourself or add a forcefield we have a modular version available. If you would like to use this, send us an e-mail.
You can now also download the latest martinize from GitHub. The major updates are always added also here below:
martinize.py and python 3 version martinize.py (version 2.6, May 12 2016)
- The option for the elastic bond lower cutoff (-el) is now correctly recognized.
- Cys bonds in gro-files and pdb-files without chain identifier are now correctly identified.
- Many, many code clean-ups and restructuring.
martinize.py (version 2.5, August 11 2015)
-Removed warnings about beta status of Martini 2.2.
-Bug fix: Fixed cases where Cys-Cys constraints were not recognized as such
martinize.py (version 2.4, August 18 2013)
-Inverted "define NO_RUBBER_BANDS" behavior.
-Changed protein backbone constraints to bonds
-Changed HIS BB-SC constraint to bonds
-Bug fix: Cys-bond length and force constant
-Bug fix: Position restraints are correctly written out when multiple chains are merged.
martinize.py (version 2.3, February 13 2013)
-Bug fix: Correctly call dssp.
-Bug fix: Correct error message when atoms are missing.
-Bug fix: Correctly merge topologies of multiple change in case of Martini 2.2P
martinize.py (version 2.2, November 27 2012)
-Added charged His to all forcefields and options to choose the His-charge state.
-Bug fix: correctly handle .gro files
-Bug fix: Correctly handle .pdb files containing hydrogens.
-Bug fix: bead types correctly set in helix starting at first residue.
-Fixed small inconsistencies in elnedyn forcefields.
-Cleaned up and added help text and warning messages.
martinize.py (version 2.1, August 23 2012)
-Bug fix: bond length in Martini 2.2 & 2.2p
-Bug fix: assignment of secondary structure
martinize.py (version 2.0, July 25 2012)
-Major clean-up and restructuring of the code
-Changed forcefield selection. Forcefield now available: Martini 2.1, Martini 2.1P, Martini 2.2, Martini 2.2P, Elnedyn, Elnedyn 2.2 and Elnedyn 2.2P
-Added function to handle new polar and charged residues in Martini 2.2P
-Several small bug fixes.
martinize-1.2.py (version 1.2, May 22th 2012)
-Fixed bug with counter in multi chain topologies.
-Corrected wrong collagen parameters.
-Fixed bug involving BBBB dihedrals in extended regions.
-Fixed bug when giving secondary structure as string.
-A test set is now available.
martinize-1.1.py (version 1.1)
-Fixed bug in pdb read-in.
-Clean up of code.
martinize-1.0.py (version 1.0)
Read more: martinize



























































































































































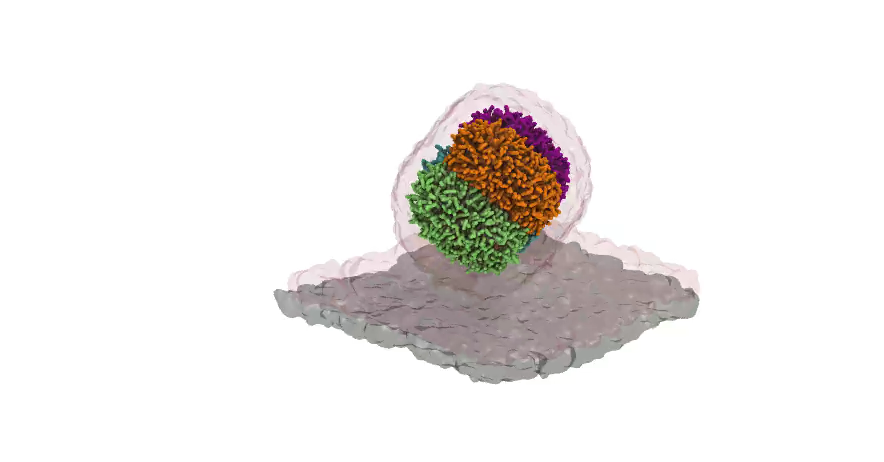
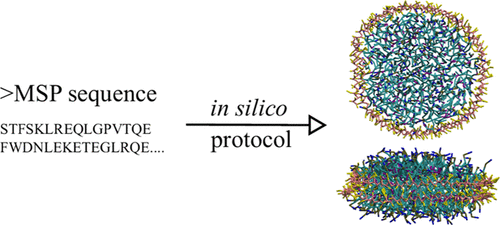 Nanodisc technology is increasingly being applied for structural and biophysical studies of membrane proteins. Here you can find a general protocol for constructing nanodiscs for molecular dynamics simulations. The protocol is written in python and based on geometric equations, making it fast and easy to modify, enabling automation and customization of nanodiscs in silico. The tool allows one to efficiently construct any membrane scaffold protein (MSP) variant given only an input sequence.
Nanodisc technology is increasingly being applied for structural and biophysical studies of membrane proteins. Here you can find a general protocol for constructing nanodiscs for molecular dynamics simulations. The protocol is written in python and based on geometric equations, making it fast and easy to modify, enabling automation and customization of nanodiscs in silico. The tool allows one to efficiently construct any membrane scaffold protein (MSP) variant given only an input sequence.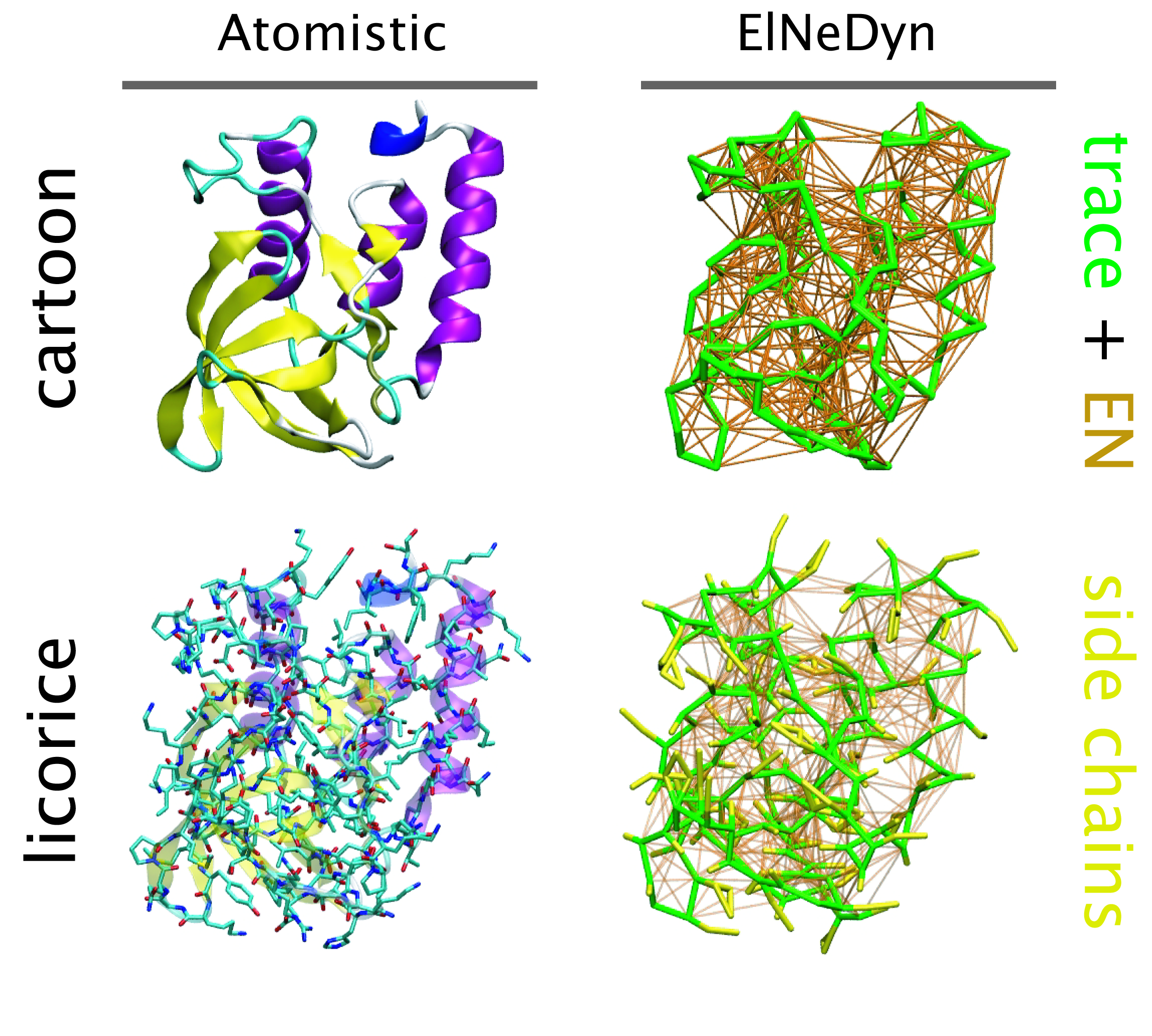 The current version of ElNeDyn focusses on modeling proteins and has been developed in conjunction with the Martini CG force field (2.0, 2.1) but can in principle be mixed with any CG model for any type of molecule. The Martini FF presents the great advantage to include a large body of biomolecules and solvents making simulations of biomolecular systems actually possible.
The current version of ElNeDyn focusses on modeling proteins and has been developed in conjunction with the Martini CG force field (2.0, 2.1) but can in principle be mixed with any CG model for any type of molecule. The Martini FF presents the great advantage to include a large body of biomolecules and solvents making simulations of biomolecular systems actually possible.