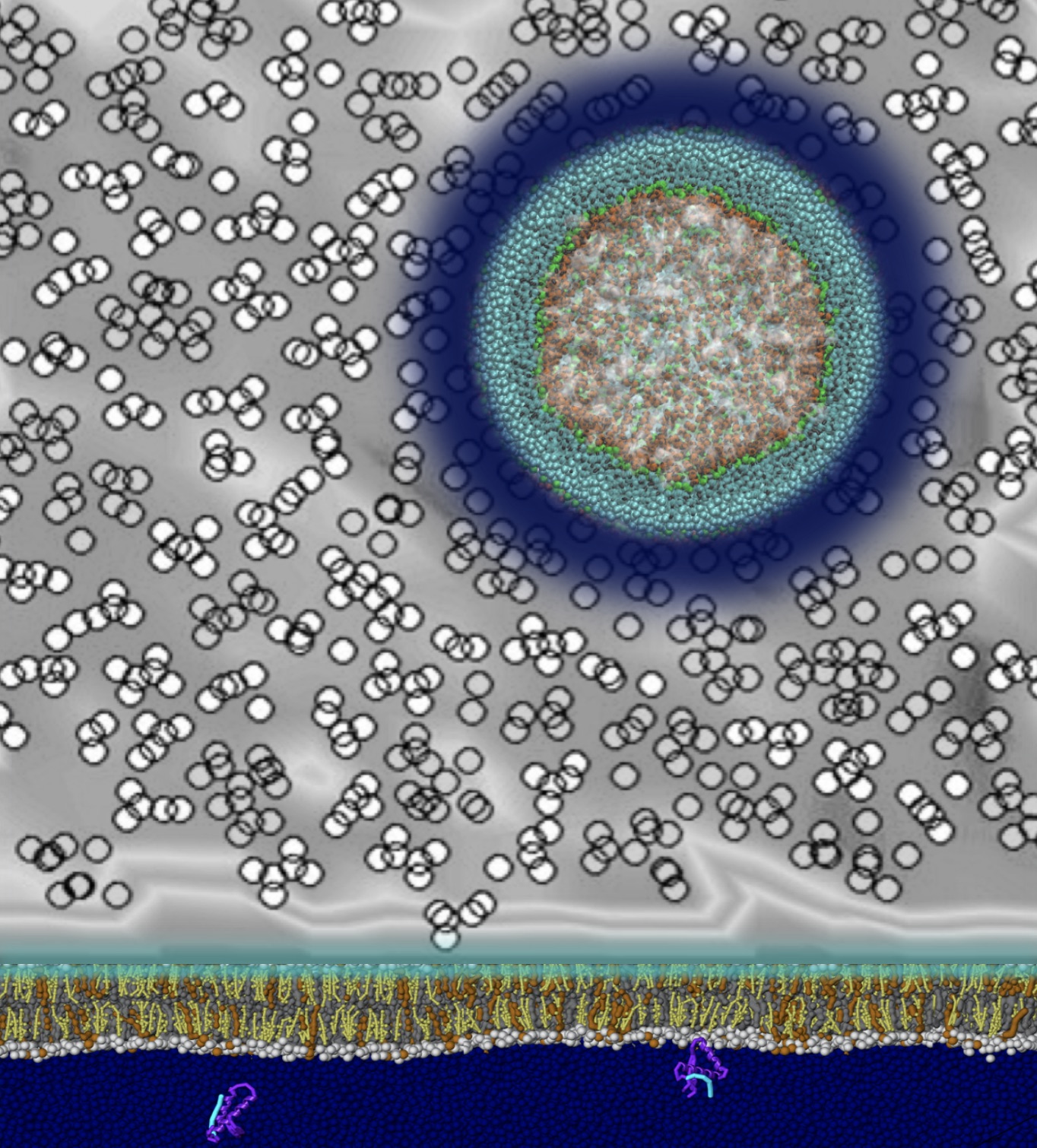
Martini Perspective in Materials Science
- Details
- Last Updated: Wednesday, 16 December 2020 11:22
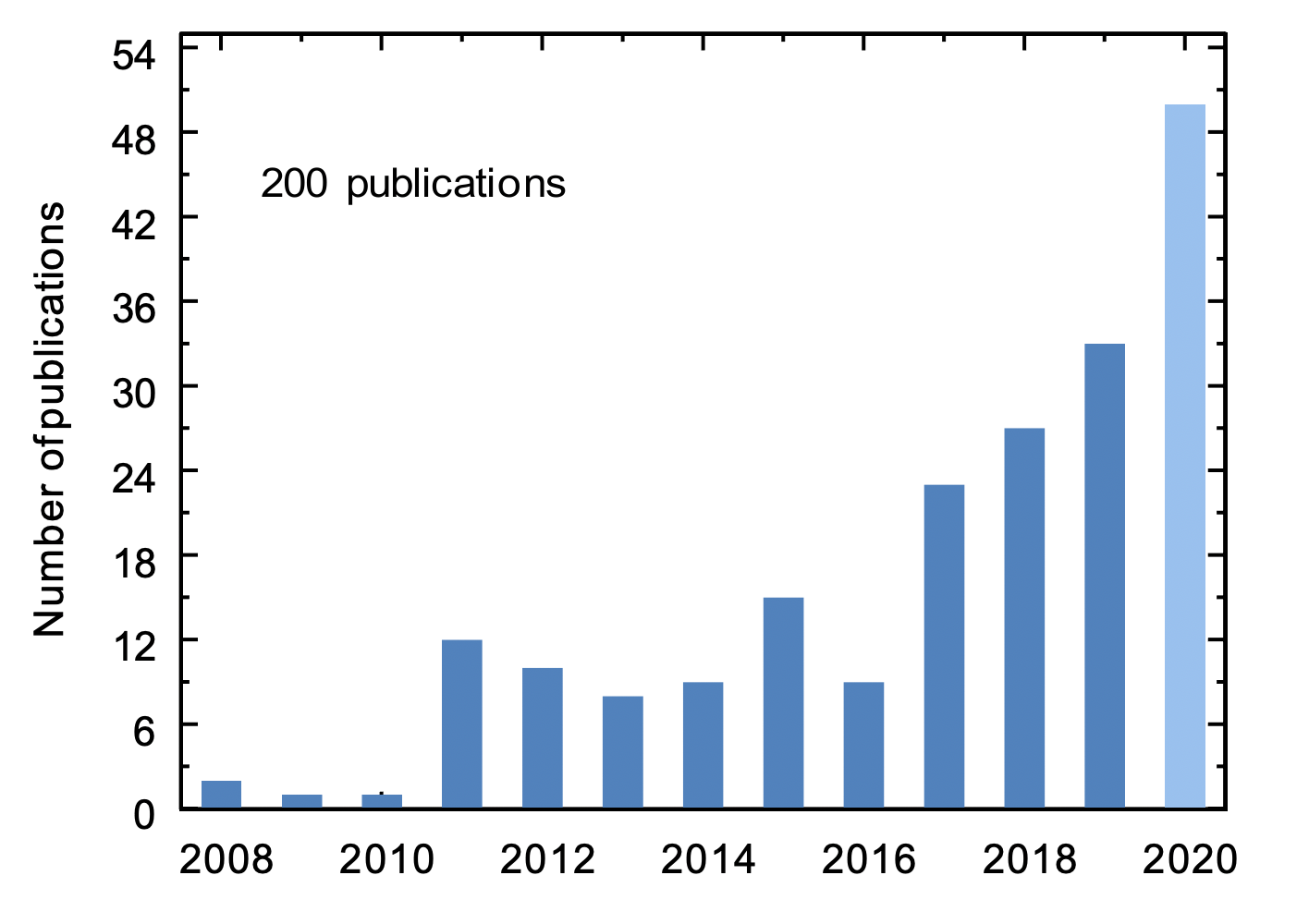
Martini applications in material science is a booming field.
We summarized the progress in this field, and provided a critical outlook, in a perspective paper which is now available in the arXiv.

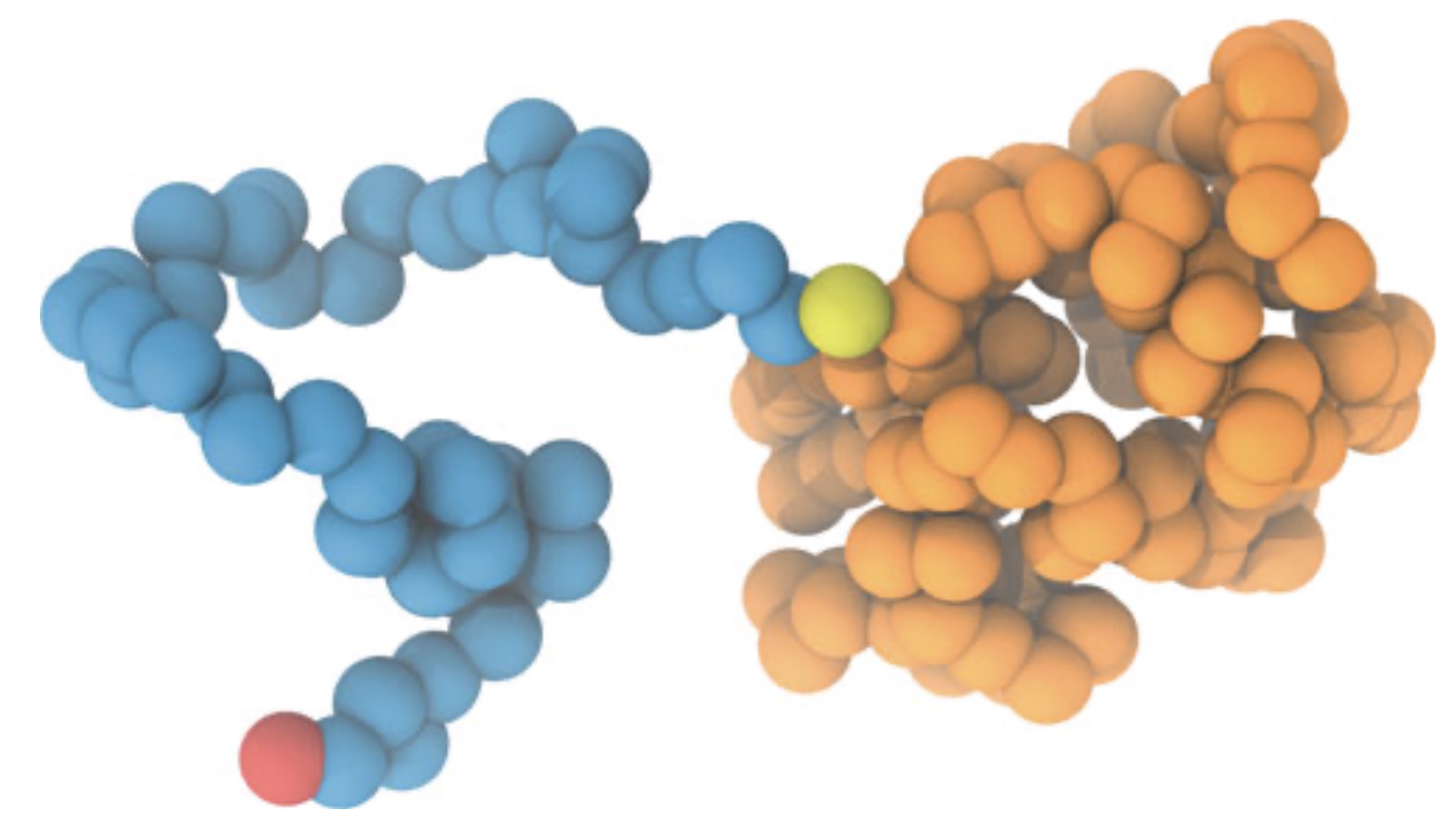 Enhancement of proteins by PEGylation is an active area of research. To gain a better insight into these interactions or even make predictions, we published a protocol on how to simulate PEGylated proteins using the latest iteration of Martini:
Enhancement of proteins by PEGylation is an active area of research. To gain a better insight into these interactions or even make predictions, we published a protocol on how to simulate PEGylated proteins using the latest iteration of Martini: